Design
An experiment of constructing viral vector.
Time and setting
Experiments were performed from May 2013 to September 2013 in the Central Laboratory, Affiliated Hospital of Qingdao University Medical College, China.
Materials
Bacterial competent cells and plasmids
Competent E. coli cells and cloning vectors GV205 were purchased from Shanghai Jikai Gene Chemical Technology Co., Ltd. and
239T cells were cryopreserved in the Center Laboratory of the Affiliated Hospital of Qingdao University Medical College, China.
Methods
Construction of cloning vector GV205
The schematic diagram of the cloning vector GV205 is shown in Figure 1.
.jpg)
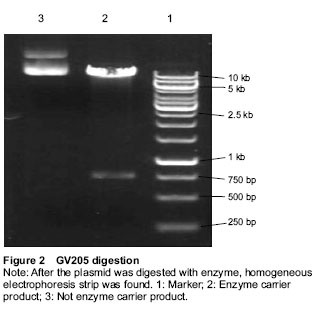
Enzyme digestion system: ddH2O 42 μL, 10 × buffer 15 μL, purified plasmid DNA (1 μg/μL) 2 μL, AgeI (5 U/μL) 1 μL, GV205 2 μg, total volume 50 μL at 37 ℃ for 2 hours. The products were electrophoresed with 1.0% agarose gel.
The carriers were identified by electrophoresis. BIRC5(5470-1)-P1 primer (Primer(+)): 5’-GAG GAT CCC CGG GTA CCG GTC GCC ACC ATG GGT GCC CCG ACG TTG C-3’; BIRC5(5470-1)-P2 primer (Primer(-)): 5’-TCA TCC TTG TAG TCG CTA TCC ATG GCA GCC AGC TGC TC-3’.
PCR reaction system: ddH2O 12.4 μL, 5 × Taq buffer 4 μL, dNTPs (2.5 mmol/L) 1.6 μL, Primer(+) (10 μmol/L) 0.4 μL, Primer(-) (10 μmol/L) 0.4 μL, Template (10 ng/μL) 1 μL, Tap polymerase 0.2 μL, total volume 20 μL.
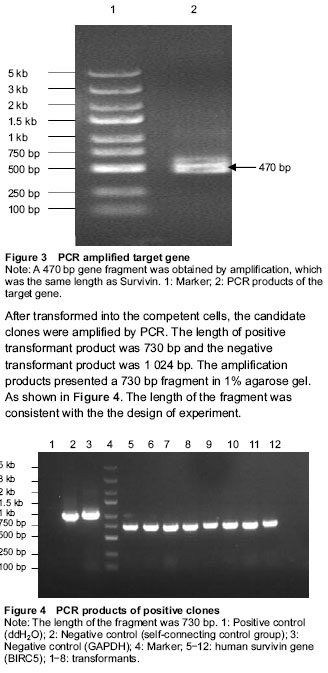
PCR conditions: First at 94 ℃ for 5 minutes; then at 94 ℃ for 30 seconds, 55 ℃ for 30 seconds, 72 ℃ for 2 minutes, total 30 cycles; last at 72 ℃ for 10 minutes; preserced at 4 ℃. The products were electrophored with 1.0% agarose gel.
Construction of recombinant plasmids
PCR products were exchanged into linear expression vector: The reagents were added according to the parameters in the following table and were conjoined by In-Fusion enzyme at 25 ℃ for 30 minutes and then at 42 ℃ for 15 minutes.
.jpg)
Transformation of competent cells: 200 μL competent cells, 1 μL plasmid and 10 μL substrate solution were added to each EP tube, and the EP tubes were placed on ice for 30 minutes. Then the tubes was transferred to a 42 ℃ preheated circulating water bath for 90 seconds, without shaking the tubes, then transferred the EP tubes to an ice bath quickly for 1-2 minutes. 800 μL Luria-Bertani medium (LB) was added to each tube, and incubated at 37 ℃ with gentle shaking for 45 minutes. 150 μL transformed competent E. coli cells were transferred to AMP resistance (100 μL/mL) of LB medium. The petri dishes were placed at room temperature until the liquid was absorbed, then petri dishes were inverted and cultured at 37 ℃ for 16 hours. The positive clones were identified by PCR.
Identifying the positive clones by PCR: Ubi-F primer: 5’-GGG TCA ATA TGT AAT TTT CAG TG-3’; pGC-E1-SEQR primer: 5’-CTA TGT TGC TCC TTT TAC GCT-3’.
PCR reaction system: ddH2O 12.4 μL, 5 × Taq buffer 4 μL, dNTPs (2.5 mmol/L) 1.6 μL, Primer(+) (10 μmol/L) 0.4 μL, Primer(-) (10 μmol/L) 0.4 μL, Template (10 ng/μL) 1 μL, Tap polymerase 0.2 μL, total volume 20 μL.
PCR conditions: First at 94 ℃ for 3 minutes; then at 94 ℃ for 30 seconds, 60 ℃ for 30 seconds, 72 ℃ for
30 seconds, total 30 cycles; last at 72 ℃ for 5 minutes; preserced at 4 ℃. The products were electrophored 1% agarose gel.
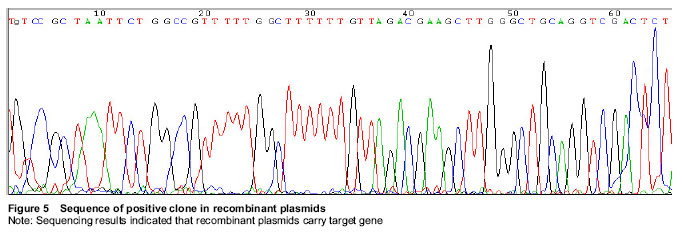
Sequencing: The positive transformants were incubated with LB medium at 37 ℃ for 16 hours, then the bacteria were preserved with glycerol and subpackaged for 200 μL/tube. The samples were sent to Shanghai Jikai Gene Chemical Technology Co., Ltd., China for sequencing.
Detection of survivin expression in the transfected 293T cells by western blot analysis
The 239T cells were recovered and cultured in DMEM medium containing 10% fetal calf serum at 37 ℃ in an atmosphere of 5% CO2, then passaged when cells reached 90% confluence.
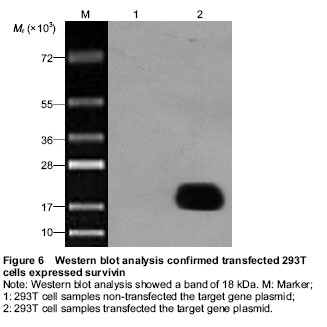
The 293T cells were digested and seeded at 4×105 per well in 24-well plates in the logarithmic phase. The Lenti-BIRC5 was transfected into the 293T cells when cells reached 80% confluence according to the Invitrogen Lipofectamine 2 000 transfection reagent manual. At 36 hours after transfection, the cells were harvested for western blot analysis.
After the original culture medium was abandoned, cells were washed with PBS twice, added with the precooled 2 × Lysis Buffer, scraped cells and transferred to EP tube, cracked cells on the ice for 15 minutes. Subsequently the cells were cracked with sonication instrument (200 W, 4 times, each time 5 seconds, at an interval of 2 seconds); centrifuged at 4 ℃, 12 000 × g for 15 minutes. The supernatant was discarded, the protein concentration was measured and adjusted for 2 μg/μL.
20 μg of each protein sample and equal volume of 2 × Loading buffer were mixed and heated at 100 ℃ for 5-
10 minutes. Then the protein sample and Marker were added into the corresponding well and were separated by SDS-PAGE at 30 mA for 2 hours.
The proteins were transferred to the PVDF membranes through the electrophoresis apparatus at 400 mA constant current and 4 ℃ for 2 hours. The PVDF membranes were blocked in 5% skim milk powder TBST at room temperature for 1 hour and incubated with the primary antibodies at room temperature for 2 hours. The membranes were then washed with TBST for three times and incubated with the secondary antibodies at room temperature for 2 hours. After final washing with TBST, the membranes were developed by using chemiluminescence according to the kit instructions of ECL-PLUS/Kit.
Main outcome measures
The recombinant lentiviral vector Lenti-BIRC5 were constructed and identified by gene sequencing. After transfected into 293T cells, the expression of recombinant lentiviral vector Flag-Survivin fusion protein was detected through western blot analysis.
.jpg)